Introduction to DwC DNA extension
Overview
Teaching: 20 min
Exercises: 10 minQuestions
What is the main purpose of the DNA Derived Data extension?
What kinds of molecular methods are supported in biodiversity data infrastructure?
If I have eDNA or metabarcoding data, how do I prepare it for public sharing?
Objectives
Explain the purpose and scope of the DNA Derived Data extension.
Distinguish between what the extension is and isn’t designed to store.
Outline the steps to prepare and map DNA data for publication.
What is the Darwin Core DNA Derived Data Extension?
The DNA Derived Data extension is a structured set of terms designed to capture information related to DNA sampling, processing, and bioinformatic methods. It incorporates terms from established genomic data standards, including Minimum Information about any (x) Sequence (MIxS), Genomic Standards Consortium (GSC), Global Genome Biodiversity Network (GGBN), and Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines for qPCR and ddPCR data.
Its purpose is to facilitate the publication of DNA related to occurrence data through biodiversity data platforms. By providing a standardized way to describe this type of data, the extension increases its usability beyond its original molecular ecology or phylogenetic context and allows it to be linked with other forms of biodiversity data, including museum specimens and field surveys.
The extension includes many fields that support methodological transparency and lab reproducibility. These fields allow users to report key experimental details such as contaminationAssessment, concentration, annealingTemp, pcr_analysis_software, and other parameters relevant to PCR conditions, quantification protocols, and data processing pipelines. This level of detail helps ensure that DNA-derived occurrence data can be interpreted, compared, and reused with confidence across studies and platforms. The current version of the extension is available here.
Introduction to Darwin Core
Need a quick refresher on Darwin Core? Check out this nice introduction.
What isn’t the DNA Derived Data Extension?
The DNA Derived Data extension is not intended for storing raw reads, full-length sequences, genome assemblies, or annotations. While it includes a
DNA_sequencefield, this is meant only for short sequences—typically DNA barcodes under ~500 base pairs. Biodiversity data platforms using this extension are not primary archives for genomic data. Instead, all comprehensive sequence data should be deposited in specialized repositories under the International Nucleotide Sequence Database Collaboration (INSDC), including NCBI and EMBL.
What about
associatedSequences?Yes, the Occurrence core includes a field called
associatedSequences, part of the MaterialEntity suite of terms. While it can be used to reference sequence data, the DNA Derived Data extension is designed to support much more detailed and structured sharing of molecular data.
History of the Extension
The DNA Derived Data extension and its accompanying guide, Publishing DNA-derived data through biodiversity data platforms, emerged in response to the growing need for clearer guidance on publishing molecular biodiversity data in a way that aligns with existing biodiversity data infrastructures. This initiative was catalyzed by conversations at the Biodiversity Next conference in 2019 and carried forward by collaborative work among GBIF, OBIS, the Biodiversity Information Standards (TDWG) Genomic Biodiversity Working Group, and the TDWG task group on Sustainable Darwin Core-MIxS interoperability.
The guide and extension were designed to reflect a consensus among DNA-focused communities about how to represent molecular methods and results in biodiversity platforms. Drawing from established standards like MIxS, GGBN, and MIQE, contributors identified terms relevant to sample collection, lab processing, and bioinformatic pipelines. These additions enable users to document essential experimental details, while maintaining interoperability with Darwin Core-based systems.
As of now, the extension is in production and actively supported on GBIF and the IPT. Meanwhile, a new data model under development may reshape how genetic data are integrated and linked across platforms in the future.
What Kinds of DNA Data Can It Handle?
The DNA Derived Data extension can be used to standardize occurrence data derived from a broad range of molecular methods, including:
- Environmental DNA (eDNA): DNA extracted directly from environmental samples (such as water, soil, or air) without isolating the source organism.
- Metabarcoding: Uses universal primers and high-throughput sequencing (HTS/NGS) to amplify and sequence specific DNA markers from mixed samples, allowing simultaneous identification of multiple taxa.
- Barcoding: Involves sequencing short, standardized DNA fragments to identify individual organisms. Often used when DNA is linked to a physical specimen or observation.
- Target capture / hybridization-based enrichment: Selectively sequences specific genomic regions across many samples, often used in phylogenomics or species delimitation.
- Whole-genome sequencing (WGS): Involves sequencing entire genomes. Though full sequences are not stored in the extension, derived occurrences based on genome assemblies or annotations can be described and linked to external repositories.
- SNP (Single Nucleotide Polymorphism) genotyping: Detects variation at specific genomic loci, commonly used in population genetics or trait association studies. Occurrence records can reflect detections, genotypes, or clustering results.
- qPCR / ddPCR (Quantitative / Droplet Digital PCR): Uses species-specific primers to detect the presence of a target organism’s DNA. These methods may not yield sequences but support presence/absence or abundance-based occurrence data.
The extension is designed to handle data derived from both individual organisms and bulk samples. It supports datasets where DNA is linked to physical material (e.g., a museum specimen, tissue, or slide) as well as those based purely on environmental or digital evidence.
What if there are fields I need that are missing?
Get involved in TDWG’s Genomic Biodiversity Interest Group! You are also welcome to post a question or issue on the Github.
Challenge: Explore the Extension
Spend about 10 minutes exploring the DNA Derived Data extension fields or browsing some occurrence records that use the extension. You can click through the available datasets on the metrics page (bottom right - click on the dataset you want to look at the occurrences for, then click to the Table tab to explore)
Which fields seem relevant to your own data? Most fields capture methodological details that often aren’t included in raw datasets—can you provide or obtain this information?
Make note of any fields you don’t recognize or fully understand. These could be good candidates for discussion or clarification.
General Approach to Using the Extension
1. Initial Checks
Before beginning the process of mapping your data, it is important to have a critical look at your dataset. Ask yourself the following questions to assess its readiness and plan your workflow:
- Is the data well organized and comprehensible? Genetic data often originates from multiple files like an OTU-table, a taxonomy table, a sample information table, and a .fasta file. You will need to understand how these relate and if they contain the necessary information (sequence and possible taxonomy for each occurrence record, sample metadata).
- Is this suitable for GBIF publication?
- Is everything crystal clear for you? Do you understand the different components of your raw data (e.g., what the “OTU-table” columns represent, how sequences link to taxonomy)?.
- Do you have the necessary metadata?
- Does the data need to be cleaned?
Metadata Fields
See OBIS’s Introduction to the EML metadata standard for a good primer.
2. Decide how your final dataset will be generally structured.
Datasets can be translated in many ways - you will find with particularly complicated datasets, there may be multiple, valid ways of mapping the data.
- Will the dataset be flat or relational? Using an extension, by definition, causes a dataset to become relational.
- Which Core element (Occurrence, Checklist or Event) is the most suitable? Currently, genetic data must be published with the Occurrence core, not the Event core. Note that a new data model is being developed which may change this in the future.
- Which Extensions are suitable for this dataset? For DNA data, the DNA Derived Data extension is essential. An extendedMeasurementsOrFact (eMoF) extension is also often used for environmental measurements or other facts associated with the sample.
- Do you have all necessary identifiers needed to link your tables? You might need to generate unique, persistent identifiers if they don’t already exist.
Available Cores and Extensions
See GBIF’s Registered Extensions for the list of registered extensions available for publication in GBIF. Remember, just because a standard is maintained in TDWG, does NOT mean it is available for use in GBIF or in other platforms.
3. Categorize Your Data
The GBIF reference guide contains recommended fields and suggestions for mapping different types of DNA data. For a guide and decision tree on determining which category your data falls into, see the Categorization of your data section.
4. Reformat your data (optional)
Genetic data is often recorded in multiple different files, and this might be the type of format received from data providers. Important data tables can include: an OTU-table, a taxonomy table, a sample information table, and a .fasta file with sequences. The OTU-table is a sequence by sample table, which records the quantity of each unique sequence found in each sample. Sequences are usually referred to by an ID, which is unique only in the dataset (e.g. asv1, asv2, asv3 …). The taxonomy table is a sequence by taxonomy table, which records the taxonomy linked to each unique sequence, as defined by the annotation method. The sample information table records the metadata of each sample (e.g. location, time, and collection method). Finally the .fasta file records the actual DNA sequence that is linked to each sequence id.
Although this data is in multiple files, each unique sequence by sample combination is considered one occurrence. Therefore the data from these tables can formatted to the “long format”, including a row for each sequence in each sample. This is an optional step, but may help with mapping to DwC.
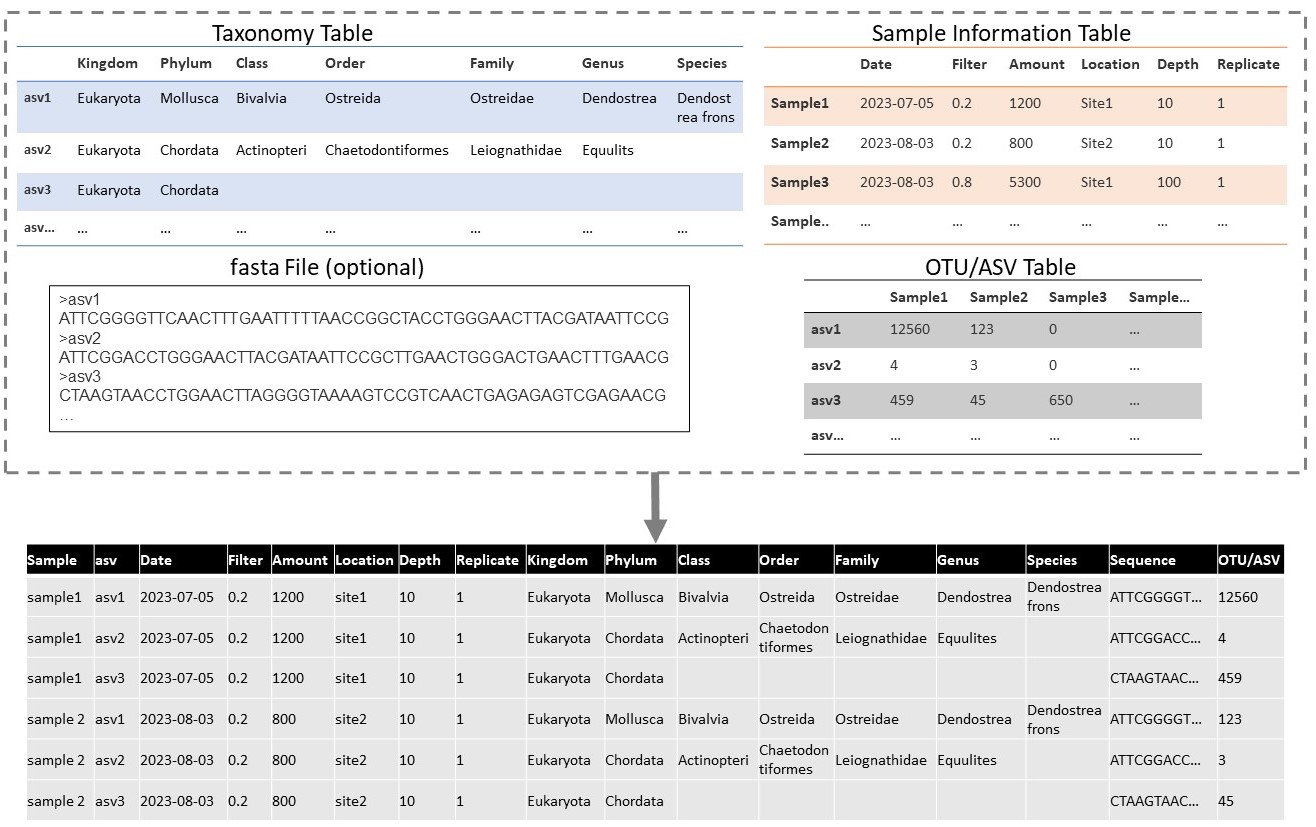
5. Conceptually Map Your Data
Before diving into the technical work of restructuring your files, take time to conceptually map your data — that is, understand how each field in your raw files will be translated into Darwin Core (DwC) terms. This step helps prevent errors and saves time by giving you a clear plan for transformation.
- Review your raw data tables (e.g., OTU-table, taxonomy table, sample metadata, FASTA file) and make a list of all fields present.
- Next, identify which DwC terms best represent each of these fields. For example:
- Taxonomic assignment →
scientificName,taxonRank,identificationRemarks - Sample metadata →
eventDate,decimalLatitude,decimalLongitude,samplingProtocol - DNA sequence →
DNA_sequence(in the DNA Derived Data extension)
- Taxonomic assignment →
- Consider relationships between tables. Use identifiers like
occurrenceID,materialSampleID, andeventIDto link records across your Occurrence Core and extensions. You may have to create your own identifiers. - Take a look through the extension fields again - Are there any that could be added that aren’t included in the data?
- Reference the GBIF mapping guide to see how others have mapped similar data types.
6. Actually Map Your Data
Once you’ve conceptually mapped your fields, the next step is to transform your data into DwC-compliant tables. This is where the actual work of data wrangling happens.
Instead of manually editing files, aim to build a scripted workflow that can:
- Import your original data tables (e.g., CSVs, TSVs, FASTA files)
- Merge and reshape them into DwC-compliant long-format tables
- Add or derive required fields (e.g., generate unique
occurrenceIDs) - Export the result as
.txtor.csvfiles suitable for DwC publication
Manual transformations are error-prone and hard to reproduce. A scripted workflow using open-source tools like R or Python ensures:
- Reproducibility: You can re-run the script when you receive new data or need to update something.
- Transparency: Others (or future you) can see exactly how the data was transformed.
- Scalability: Scripts handle large datasets more easily than spreadsheets.
Examples and Resources
- See this list of R Notebooks for examples of these scripts.
- A full example workflow for eDNA data is available here.
- See also Reyserhove et al. 2020 for a published checklist recipe for creating a reproducible workflow.
Key Points
The DNA Derived Data extension standardizes how DNA-based occurrence data is shared through biodiversity platforms like GBIF.
It supports detailed reporting of lab and bioinformatic methods, enhancing reproducibility and data reuse.
It is not for storing raw reads or full genome sequences—only short sequences like barcodes.
The extension builds on established standards (MIxS, GGBN, MIQE) and integrates with Darwin Core using the Occurrence Core.