Introduction to DNA Data
Overview
Teaching: 45 min
Exercises: 0 minQuestions
Where can I find examples of DNA-derived data being published?
How do BOLD, GenBank, and GBIF interact?
What are the categories of DNA-derived data?
Objectives
Review examples of DNA-derived data published to GBIF, BOLD, and GenBank.
Understand how DNA data are categorized and shared between platforms.
Learn why publishing imperfect data is still valuable.
Explore how BOLD, GenBank, and GBIF interact and share data.
Categories of DNA Data
DNA derived data are increasingly being used to document taxon occurrences. This genetic data may come from a sampling event, an individual organism, may be linked to physical material (or not), or may result from DNA detection methods e.g., metabarcoding or qPCR. Thus genetic data may reflect a single organism, or may include information from bulk samples with many individuals. Still, DNA-derived occurrence data of species should be documented as standardized and as reproducible as possible.
To ensure DNA data are useful to the broadest possible community, a community guide entitled Publishing DNA-derived data through biodiversity data platforms was published by GBIF, OBIS, and others. This guide is supported by the DNA derived data extension for Darwin Core, which incorporates MIxS terms into the Darwin Core standard. There are 5 categories for which genetic data could fall into:
-
DNA-derived occurrences
These are occurrence records directly based on the detection of DNA in an environmental sample. Examples include environmental DNA (eDNA), metabarcoding, and metagenomics. In these cases, the presence of a taxon is inferred from sequence data rather than direct observation or capture of the organism itself. -
Enriched occurrences
These involve individual specimens that have been sequenced, such as through DNA barcoding, genome, or transcriptome projects. Typically, these records are linked to a vouchered specimen or observed organism, enriching the original occurrence record. -
Targeted species detection
This refers to the use of molecular methods designed to detect a specific species—often in a presence/absence fashion. For example, qPCR/ddPCR assays for specific species or pathogens. These detections may or may not be linked to physical samples, but the assays themselves are often well documented. -
Name references
These involve taxonomic names derived from sequence clusters, such as Operational Taxonomic Units (OTUs), Amplicon Sequence Variants (ASVs), or Barcode Index Numbers (BINs). While not direct occurrences, these names are often used in ecological checklists or in reference to taxonomic hypotheses generated from DNA. -
Metadata only
Some datasets may not contain data at all but describe metadata in broad terms, such as project authors, scope or laboratory workflows and sequencing methods. These datasets can still be valuable, especially when paired with future data or linked through identifiers.
Categorizing Your DNA Data
For a guide and decision tree on determining which category your DNA data falls into, see the Data packaging and mapping section of the GBIF guide.
Workshop Focus
For the purpose of this workshop, we will focus on the first three categories:
DNA-derived occurrences, Enriched occurrences, and Targeted species detection.
These represent the most common and practical entry points for publishing DNA data to biodiversity platforms like GBIF.
Overview of DNA-derived Occurrences and Detection Data
Overview of Enriched Occurrences
If genetic material is connected to an observation or a physical sample, we call it an “enriched occurrence.” This means the DNA isn’t the only line of evidence—we can also trace the data back to a specific organism that was seen, collected, or otherwise documented.
This category includes a range of sequencing approaches such as DNA barcoding, whole genome sequencing, transcriptomics, target capture, or restriction digest assays (e.g., RAD-seq or ddRAD), as long as the sequences can be clearly associated with a reference sample (e.g., a museum specimen, tissue archive, or observational record). In many cases, metadata like voucher numbers, collection information, and taxonomic IDs are available and critical for data reuse.
Importantly, enriched occurrences can be derived from both newly collected material and historical specimens. In some instances, an entire physical specimen may not have been retained—such as when a sample (e.g., hair, feathers, or plant tissue) was taken in the field—but sufficient metadata (e.g., date, location, taxon identification) still allows the genetic data to be linked to a valid occurrence record.
Introduction to NCBI and BOLD
BOLD (Barcode of Life Data Systems)
The International Barcode of Life (iBOL) consortium has developed the Barcode of Life Data Systems (BOLD), an informatics platform that includes barcode sequences from specimens held in natural history collections around the world. Consisting of four main modules, a data portal, an educational portal, a registry of BINs (putative species), and a data collection and analysis workbench, it supports the acquisition, storage, analysis, and publication of DNA barcode records. BOLD’s primary purpose is to provide a centralized resource for DNA barcoding, which is a method used for species identification through DNA sequences, typically from the mitochondrial cytochrome c oxidase subunit 1 (COI) gene.
Developed at the Canadian Centre for DNA Barcoding (CCDB) at the University of Guelph in Canada in response to the BARCODE 500K project, BOLD is widely used in the DNA barcoding community and is the standard platform through which data are typically returned when samples are processed by CCDB.
NCBI
National Center for Biotechnology Information (NCBI) houses a series of interconnected databases relevant to biotechnology and biomedicine and is an important resource for bioinformatics tools and services. It maintains many databases, storing information such as DNA and protein sequences, genomes, protein structures and even a taxonomic database.
GenBank is a comprehensive public database of nucleotide sequences and supporting bibliographic and biological annotations. NCBI builds GenBank primarily from submissions of sequence data from authors and from bulk submissions of whole-genome shotgun (WGS) and other high-throughput data from sequencing centers. GenBank is accessible through the NCBI Nucleotide database, which links records to related records in other databases such as taxonomy, genomes, protein sequences and structures, and biomedical journal literature in the PubMed database.
Although Sanger sequencing—a first-generation, cost-effective method for generating short reads—is still commonly used for DNA barcoding and other targeted applications, many researchers now rely on next-generation (e.g., Illumina) and third-generation (e.g., PacBio, Oxford Nanopore) sequencing technologies for high-throughput studies of genomes, transcriptomes, and metagenomes. Raw sequence data from these platforms are typically submitted to the Sequence Read Archive (SRA), while downstream products such as genome assemblies and annotations are deposited in the dedicated Genome database. Annotated individual Genes and Proteins are submitted to their respective repositories, ensuring the broader accessibility and reuse of these data across research communities.
Interestingly, the BioSample database stores descriptive metadata about the biological materials used to generate data submitted to NCBI’s primary archives. These materials often include cell lines, tissue samples, or environmental isolates, but they can also represent individual organisms. Despite this overlap, BioSample records remain largely disconnected from biodiversity platforms like GBIF—highlighting a potential area for future integration and exploration.
Rabbit Hole
The BioSample database offers three attribute “packages” that may be especially relevant for museum specimens: animal, plant, and invertebrate. Each includes a
specimen_voucherfield, which serves as a reference to the physical specimen used in the analysis.specimen_voucher:
“Identifier for the physical specimen. Use the format:"[<institution-code>:[<collection-code>:]]<specimen_id>", e.g.,UAM:Mamm:52179. Intended as a reference to the physical specimen that remains after it was analyzed. If the specimen was destroyed in the process of analysis, electronic images (e-vouchers) are an adequate substitute for a physical voucher specimen. Ideally the specimens will be deposited in a curated museum, herbarium, or frozen tissue collection, but often they will remain in a personal or laboratory collection for some time before they are deposited in a curated collection. There are three forms of specimen_voucher qualifiers. If the text of the qualifier includes one or more colons it is a ‘structured voucher’. Structured vouchers include institution-codes (and optional collection-codes) taken from a controlled vocabulary maintained by the INSDC that denotes the museum or herbarium collection where the specimen resides, please visit: http://www.insdc.org/controlled-vocabulary-specimenvoucher-qualifier.’”
- Browse the list of recognized collections: coll_dump.txt
- Explore existing records using
specimen_voucher: NCBI nucleotide search
Do These Platforms Share Data?
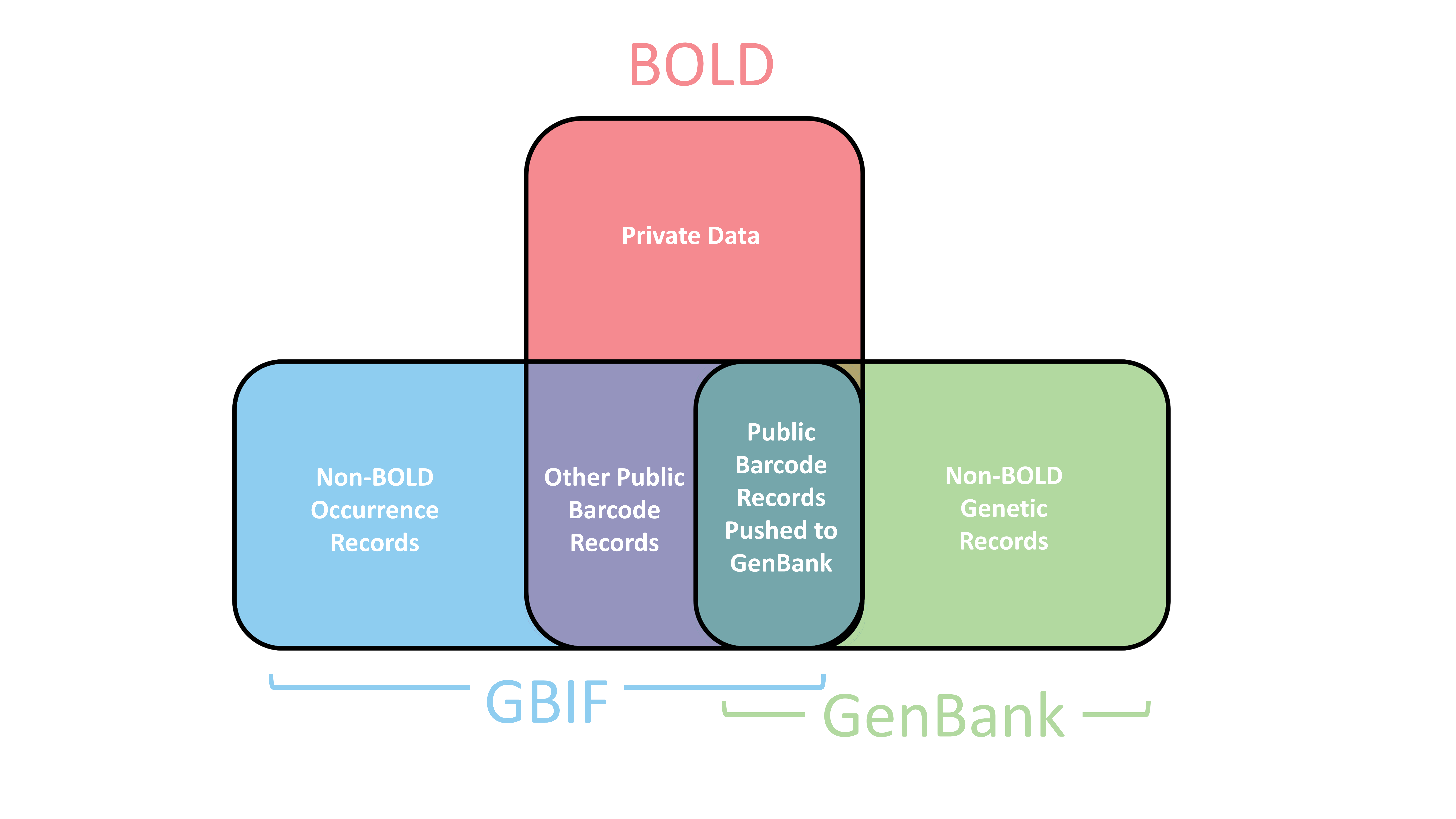
Yes—at least in part. The relationships between BOLD, GenBank, and GBIF involve some data sharing, but they serve different roles and audiences, they do not share all of their data with each other, nor are they consistently cross-referenced or synced.
- BOLD: Data in BOLD are categorized as either public or private. Only data marked as public (or those under embargo) can be pushed to GenBank, allowing for automatic submission of data upon initiation by the data manager. Once submitted, records are linked and updated from BOLD to GenBank, with taxonomic identification changes automatically communicated. See the handbook for more information.
- GenBank: Stores a wide variety of DNA sequence data, including records submitted from BOLD. However, data is not automatically shared outward from GenBank, nor is it typically ingested by BOLD or GBIF.
- GBIF: Ingests all publicly available data from BOLD on a weekly basis and makes it accessible as part of global biodiversity occurrence data.
Same Record, Different Platforms
To see how the same DNA record can be distributed across platforms, explore this example from the Florida Museum:
It’s ok to not have it all figured out yet
Publishing biodiversity data can be messy—and that’s perfectly okay. A good example is how NEON data currently appear in BOLD and GBIF. Sometimes, related datasets—like fish vouchers and the DNA extracted from those same fish—end up split across datasets instead of being linked as one record.
For instance, a mammal voucher collected by NEON is published to GBIF, while the DNA material from that mammal is stored separately in a different dataset, also published to GBIF. Because this DNA material was sequenced, it is available on BOLD, which is also published to GBIF. This has resulted in three separate records, which should probably only be one. Also note the taxonomic discrepencies!
These kinds of disconnects are common. The reality is: aligning genetic, voucher, and occurrence data takes time, tools, and coordination—and not every project has those resources. But that doesn’t mean the data shouldn’t be shared.
Even if it’s imperfect, getting the data out there helps move science forward. It’s far better than letting valuable information sit hidden for years, waiting for everything to be “just right.”
Good enough and accessible beats perfect and invisible.
 Three GBIF records representing what is the same NEON specimen. a) shows the mammal voucher; b) is the DNA material derived from that voucher; and c) is the DNA barcode record from BOLD, which is also pushed to GBIF. These records are not linked, despite being biologically connected.
Three GBIF records representing what is the same NEON specimen. a) shows the mammal voucher; b) is the DNA material derived from that voucher; and c) is the DNA barcode record from BOLD, which is also pushed to GBIF. These records are not linked, despite being biologically connected.
Key Points
DNA data can be shared on BOLD, GenBank, and GBIF, with varying levels of interaction.
There are five main categories for DNA data, including DNA-derived occurrences and enriched occurrences.
Even if data isn’t perfect, making it accessible can lead to valuable insights.
Data discrepancies, like taxonomic errors, are common but don’t prevent data from being useful.